Our research interests. For details, click panels.
Atomic Scale Simulation
Microkinetics
Reactor Simulation
Machine Learning
Atomic Scale Simulation
Summary
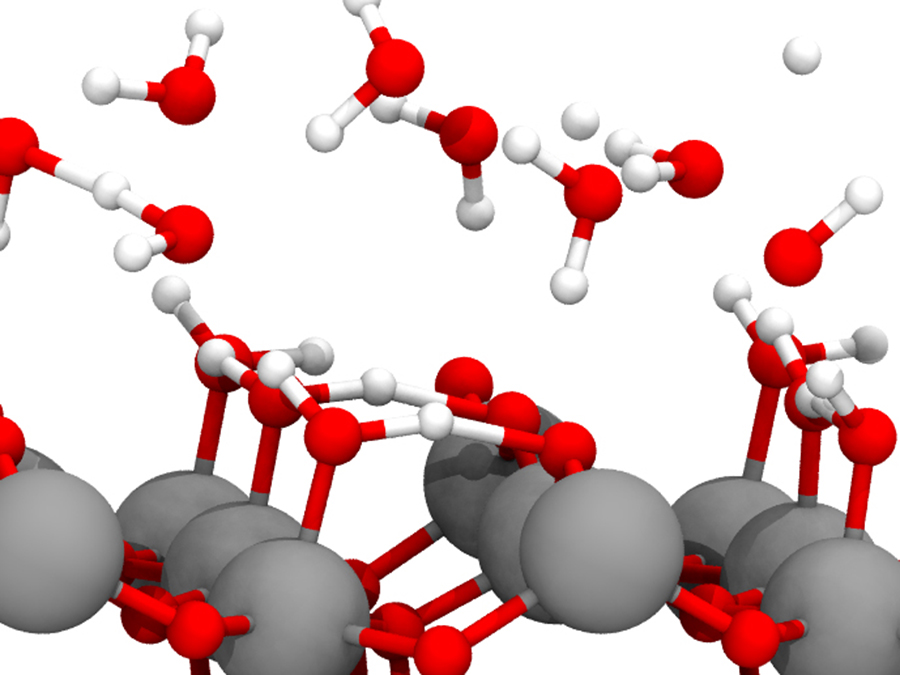
The behavior of atoms and molecules are governed by Schrodinger equation, a quantum mechanical fundamental equation. Then, one can understand or predict the atomic/molecular behavior by solving the Schrodinger equation. Simulation techniques based on this idea is called first-principle simulation, quantum chemical simulation, or ab initio simulation.
In our group, we apply this approach to materials such as catalysts, batteries, fuel cells, etc. Using first-principle simulation, one can predict the quantities like reaction heat, activation energy, etc. for the chemical reaction. These are important quantity to know whether chemical reactions undergo or not.
Research outcomes
CH4 activation reaction on Ga2O3 surface (collaboration with Sugimoto group in IMS):
Comm. Chem. (2023)
NH3 electrosynthesis on Ru/BaCeO3 catalyst (collaboration with Otomo group in TITech):
ACS Omega (2022)
On-surface porphyrin synthesis and its analysis by scanning tunneling microscopy and theoretical calculation (collaboration with Kawai group in NIMS):
Angew. Chem. (2022)
Microkinetics
Summary
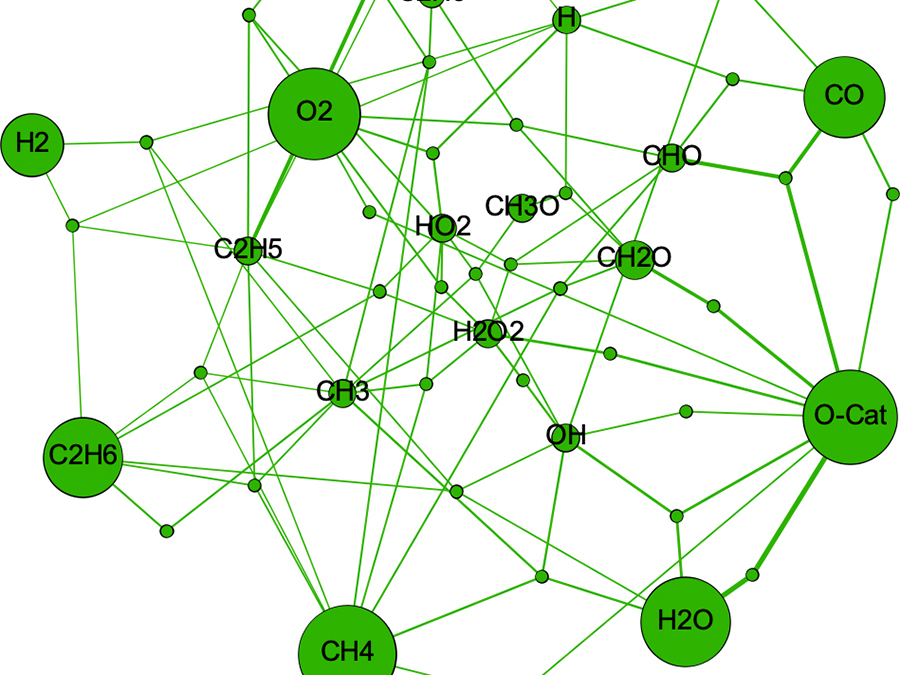
The purpose of catalytic reactions is to convert chemical compounds. For this this purpose, the quantities called conversion (how much of the raw material has reacted) or selectivity (how much the target product is obtained among several product species) are important. If these quantities can be predicted from theoretical calculations alone, there is no need to conduct numerous experiments, thereby reducing the human and environmental burden. However, to calculate conversion and selectivity, it is not sufficient to consider only one elementary reaction; it is necessary to consider many elementary reactions at the same time. Moreover, it is necessary to know the parameters such as rate constants for all elementary reaction; it is not easy task for experimentalists.
Therefore, we focus on "microkinetics" in which kinetic parameters for each elementary reaction are calculated by first-principles calculations and used to solve kinetic equations consisting of a large number of elementary reactions.
Once the reaction rate of each compound is determined by methods such as microkinetics, it is possible to simulate the whole chemical reactor in which the chemical reaction takes place. To do this, one needs to consider additional physics or chemistry such as material flow, heat flow, etc. This type of calculation is done with a method called computational fluid dynamics (CFD), and CFD enables realistic simulation of chemical reactions.
In our laboratory, we will challenge these problems using CFD software such as COMSOL and openFOAM.
Machine Learning
Summary
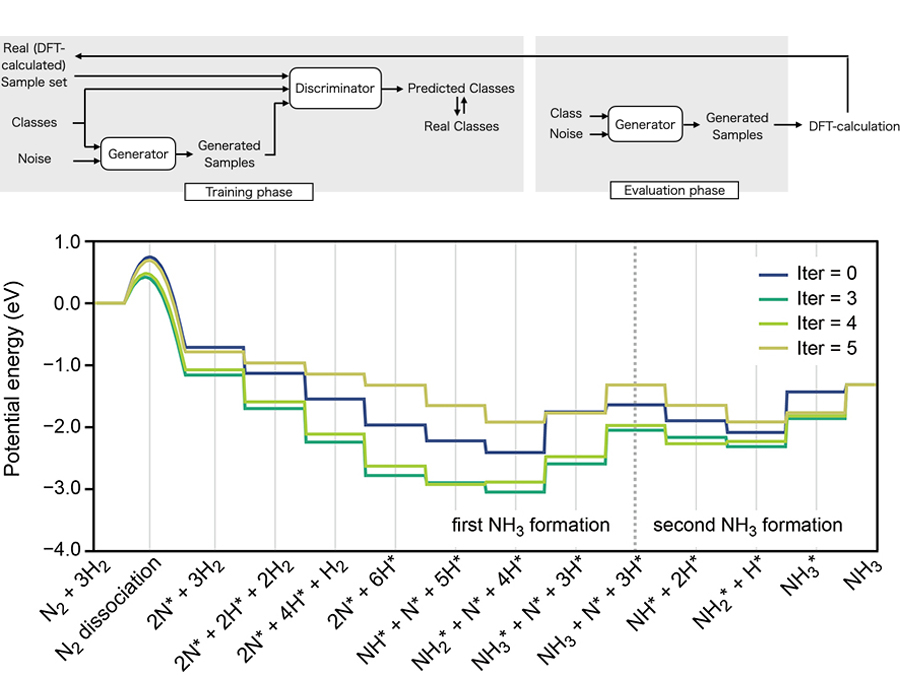
It is possible to explain the high performance of a material using first-principles calculations or reaction kinetics. On the other hand, it is not easy to use that knowledge to propose brand-new materials. Machine learning is one method that makes this possible. In our laboratory, we mainly focus on catalysts, batteries, and fuel cells, generating data by theoretical simulations and analyzing the results by regression and clustering, or proposing new materials through neural networks.
Research outcomes
Heterogeneous catalyst design by first-principle calculation and generative adversarial network:
Sci. Rep. (2022)
Search for a lithium ion battery electrolyte with quantum chemistry calculation and sparse modeling:
PCCP (2019)