研究対象
当研究室での研究対象です。詳細は各パネルをクリックしてください。
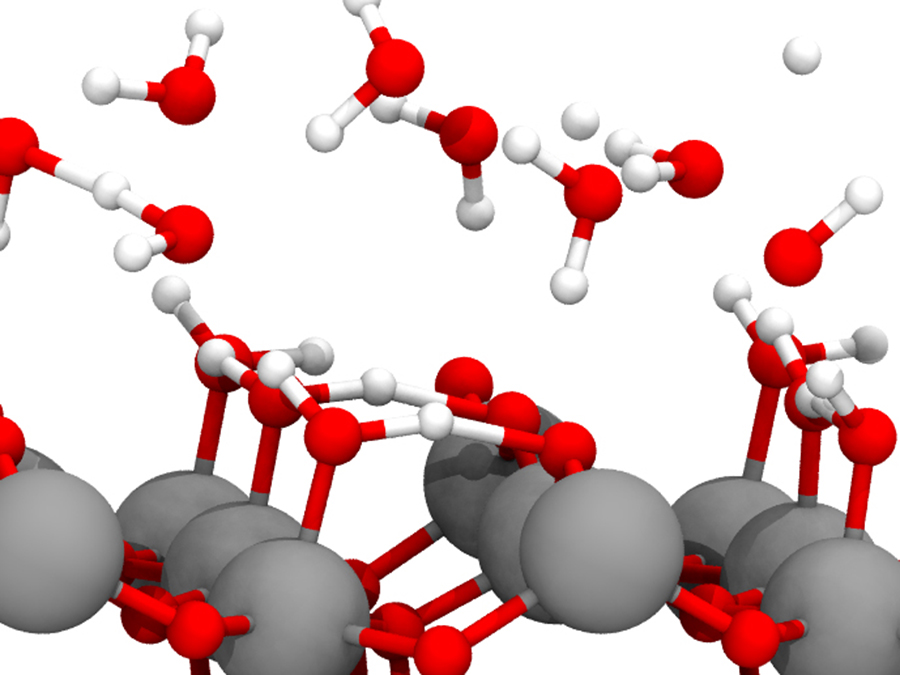
原子スケールの理論・計算化学
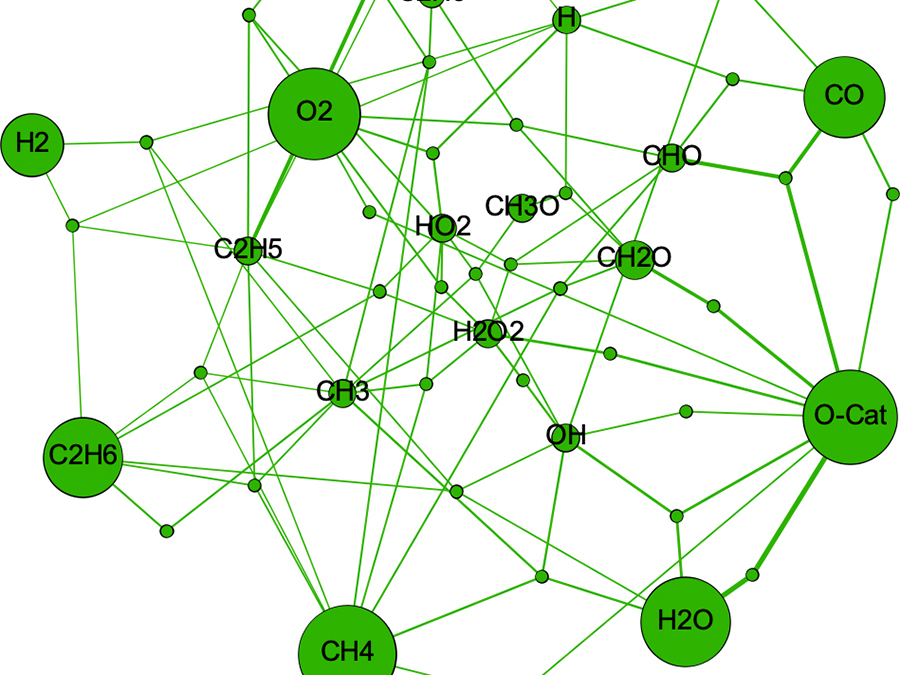
微視的反応速度論

反応器シミュレーション
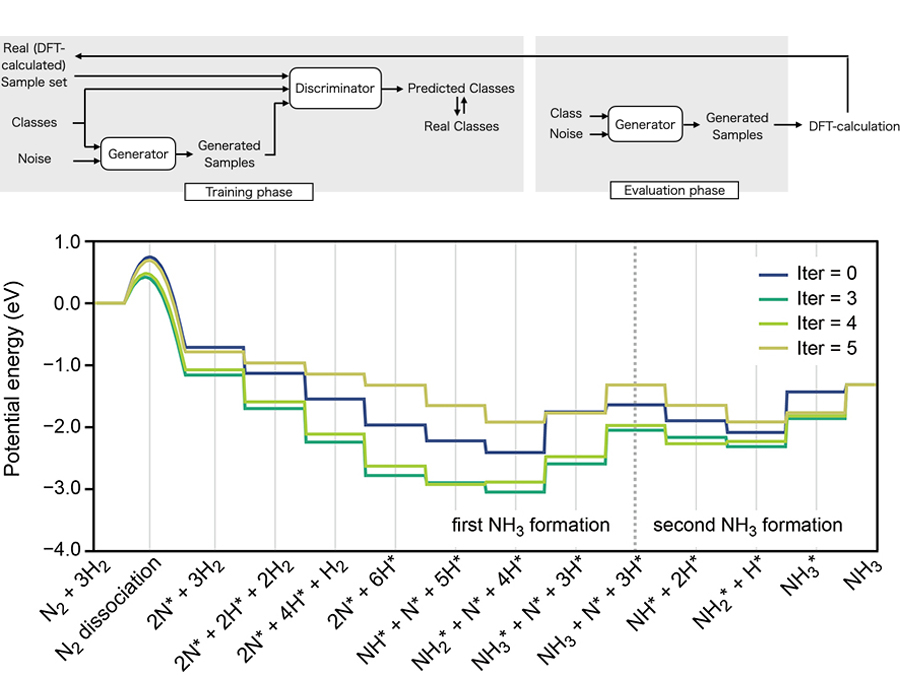
機械学習